Theory
Full stack materials design requires understanding the relationship between fine-grained chemical detail and resulting macroscopic properties. The computational workflow at the BioPacific MIP provides a suite of tools to do exactly that.
There are three levels of simulation capabilities in toolbox: all-atom resolution molecular dynamics, coarse-grained molecular dynamics, and field theoretic simulations. The key to making this all work is the careful construction of coarse-grained molecular models that are simultaneously simulatable in field theoretic simulations while as faithful as possible to the underlying all atom molecular dynamics simulations.
Many of the details, nuances, and theoretical considerations of model selection are outlined in the best practices page.
On this page we give a high level overview of the mathematical transformations underlying the development of coarse grained models from all atom simulations, and the field theoretic, continuum simulation of particle based models.
From all atom to coarse-grained models: the relative entropy
Systematic bottom-up coarse-graining refers to the systematic elimination of chemical degrees of freedom. Most commonly, this is effected by replacing collections of atoms with pseudo-atoms, or beads, typically representing the center of mass of the atoms being replaced.
Given a particular mapping from all atom coordinates \(\{\mathbf{R}\}\) to coarse grained coordinates \(\{\mathbf{r}\}\), the challenge is to determine a suitable interaction potential between coarse-grained beads that reproduces characteristics of the all atom simulation. Formally, this means that instead of simulating with interaction energy \(U(\{\mathbf{r}\})\) governing the all atom representation, one needs to find an appropriate potential of mean force \(W(\{\mathbf{R}\})\).
The optimal potential of mean force \(W\) exactly reproducing configurational distributions of the all atom model can be formally defined:
$$ \beta W(\{\mathbf{R}\}) = -\ln\left(V^{N-n} \int d\{\mathbf{r}\} e^{-H(\{\mathbf{r}\})} \delta(M(\{\mathbf{r}\})-\{\mathbf{R}\}) \right) $$ where \(M(\\{\mathbf{r}\\})\) is a mapping operator converting all-atom coordinates to coarse-grained coordinates.
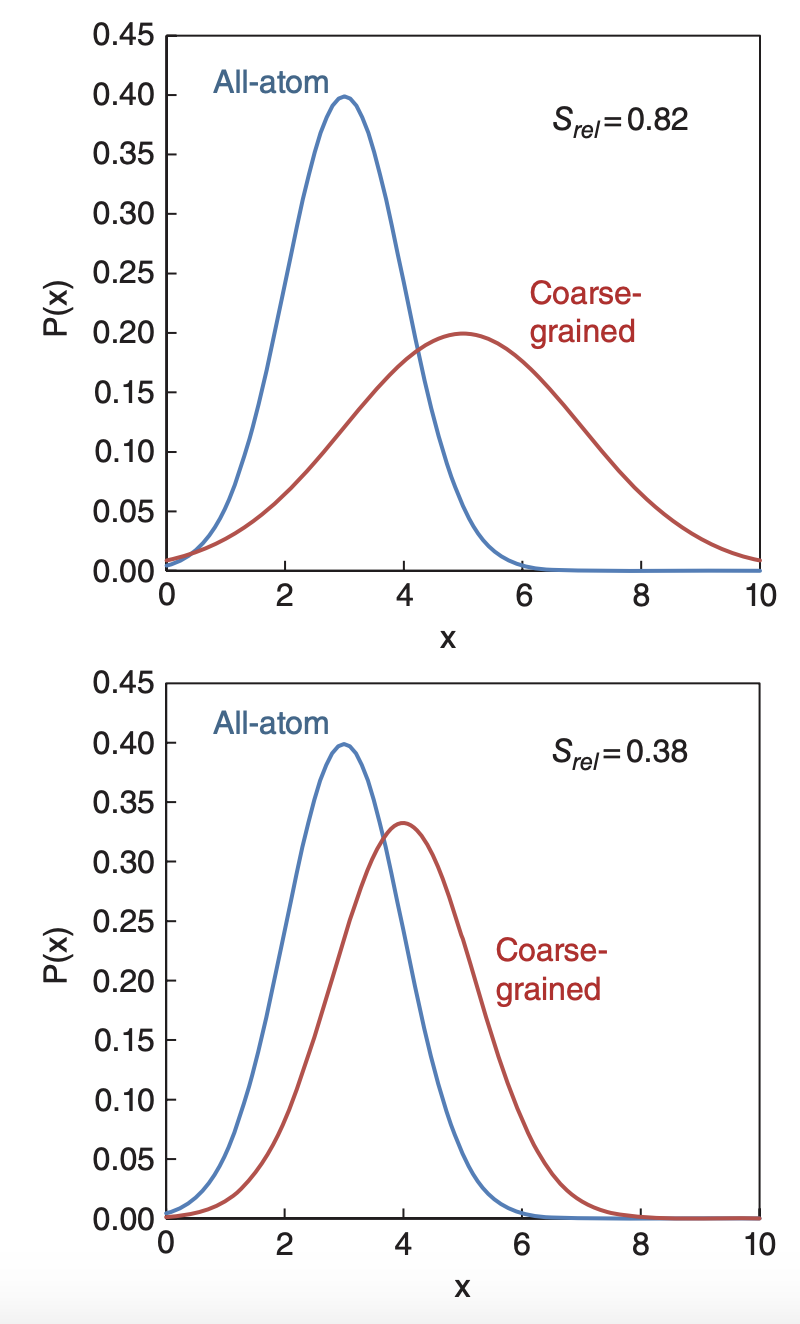
However, this object is multi-body in nature and intractable to determine. In practice, pairwise potentials are more practical, and several procedures exist to find pairwise approximations of \(W\). The coarse graining technique employed in the BioPACIFIC simulation software suite is relative entropy minimization, which seeks to minimize the information loss, or maximize the configurational probability distribution overlap between the coarse-grained system and (mapped) all atom model, as measured by the relative entropy between two probability distributions \(P_1(x)\) and \(P_2(x)\):
Schematically, in 1D, the relative entropy can be visualized as thus (image taken from Shell, Adv. Chem. Phys. 2016):
In practice, the minimization of the relative entropy can become a high dimensional optimization problem achieved by solving for:
where \(\lambda_i\) refer to coarse grained interaction parameters, such as interaction coefficients or interaction ranges.
This is handled by the in-house coarse graining simulation software at BioPacific MIP.
From coarse-grained particle models to continuum: field theoretic models
There are several exact mathematical transformations from particle to continuum descriptions. The one employed in the BioPACIFIC simulation software suite is the auxiliary field approach.
A good series of lectures on field theory can be found courtesy of Professor Rob Riggleman.
In short, the first step is to define particle density \(\rho_\alpha\) of \(\alpha\)-species beads as such:
where the sum is over all particles in the system of species \(\alpha\). It is also common to smear these densities by replacing the \(\delta\) function with a Gaussian \(\Gamma\):
This then allows for the critical mathematical trick of using the Hubbard Stratonovich transformation, an exact transformation that allows for the transformation of any pairwise, positive definite interaction \(U(r,r')\).
Definint the shorthand: $$\begin{eqnarray} \langle f | A | f \rangle \equiv& \int_{dr}\int_{dr'}f(r)A(r,r')f(r') \end{eqnarray} $$ $$ \langle f, g \rangle \equiv \int_{dr}f(r)g(r) $$
we express the Hubbard Stratonovich transformation: $$\begin{eqnarray} \exp\bigg(-\frac12\langle \rho | U | \rho \rangle\bigg) =& \frac{1}{Z}\int D\omega \exp\bigg(-\frac12\langle \omega | U | \omega \rangle+i\langle \rho, \omega \rangle\bigg) \end{eqnarray} $$ $$ Z = \int D\omega \exp\left(-\frac12\ \langle\omega | U | \omega\rangle\right) $$
where we have introduced functional integrals over a newly introduced "auxiliary" field \(\omega\).
Note that the density fields only appear linearly in the argument of the exponential, or the new "effective Hamiltonian". This decouples all configuration integrals from one another and is what allows field theories to efficiently treate the configurational space of large molecules. Analytic continuation of the functional integral over \(\omega\) into the complex plane allows for efficient numerical evaluation of the field integral by concentrating sampling around saddle points. Numerical techniques for solving this formulation are described in literature.
The field theory enables full field theoretic simulation and useful approximations like self-consistent field theory. It also provides helpful analytical tools for understanding polymeric behavior in disordered solutions, using mean-field analysis and the random phase approximation.